
今回の記事は下記のような読者に向けて書いています。
「pythonを活用してケモインフォマティクスを実践したい」
「rdkitで何ができるか知りたい」
「rdkitを使ったケモインフォマティクスの実施例を見てみたい」
pythonを活用したケモインフォマティクスでは、『rdkit』はマストなので是非この記事で使い方を理解してもらえればと思います。
rdkitでできること

rdkitを使用することで、「pythonで化学構造を取り扱える」ようになります。
rdkitを活用することで下記例のような事ができます。
・部分構造検索
・化学構造の自動生成
今回の記事では特に、「化学構造のベクトル化」について解説します。
化学構造をベクトルに変換することで、機械学習で化学構造を取り扱えるようになります。
つまり、化学構造を取り扱う機械学習(ケモインフォマティクス)を実践できるようになります。
✅ rdkitについてより詳細まで知りたい方は「化学の新しいカタチ」さん記事参照。

早速、rdkitを用いた「化学構造のベクトル化」について解説します。
化学構造のベクトル化
化学構造のベクトル化は下記の流れで行います。
『SMILES→molファイル→ベクトル』の流れで、化学構造をベクトルに変換(機械学習で使える形に変換)しているんだ〜くらいの理解度で大丈夫です。
それぞれのステップを解説します。
まずは『SMILES→molファイル』への変換です。
SMILES→molファイル
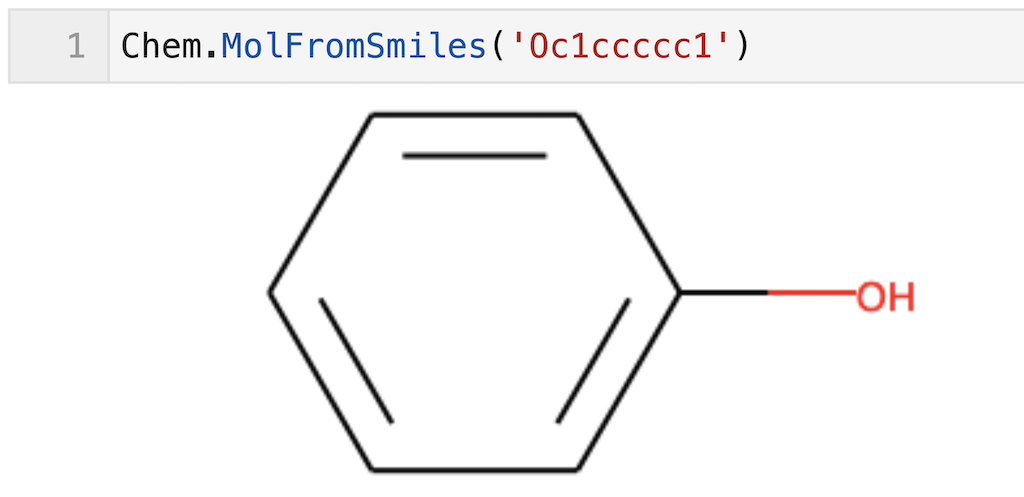
SMILES→molファイルへの変換は、Chem.MolFromSmiles(化合物のSMILES)を使えばいい。
フェノールをmolファイルに変換する例を下記に示す。molファイルを出力すると、フェノール(化学構造)が出力されます。
from rdkit import Chem
Chem.MolFromSmiles('Oc1ccccc1')
次に『molファイル→ベクトル』に変換する手法について解説します。
molファイル→ベクトル
molファイル→ベクトルに変換する手法はいくつかあります。
・分子記述子(rdkit記述子、mordred記述子)
フィンガープリントはベクトルの数値が「0 or 1」で表現されるのに対して、記述子は「数値」で表現されます。
機械学習では、どの手法を使えば予測精度が高くなるのかわからないので、色々試して、予測精度が高い手法を選択すればいいと思います。
長々と書きましたが、rdkitを活用して、「smiles→molファイル→ベクトル」の流れで、化学構造をベクトルに変換(機械学習で使える形に変換)しているんだ〜くらいの理解度でOKです。
そして化学構造をベクトルに変換できれば、あとはいつもの機械学習と同じです。
次に実際にrdkitを使ってケモインフォマティクスを実践します。
✅ rdkitの環境構築がまだの人は下記記事参照。
anacondaで仮想環境を構築してrdkitが使える環境を構築しています。

それではここからは実際にケモインフォマティクスを実践していきます。
rdkitを使ったケモインフォマティクスの実践

今回は、rdkitを使って水溶解度予測モデルを構築します。
目的変数を水溶解度、説明変数を化学構造(ベクトル)として、その2つを結ぶFを機械学習で構築します。
目的変数の水溶解度は、水溶解度をS(mol/L)とすると、対数変換したlog10Sの形で使用します。
まずはデータを読み込みます。
データの読み込み
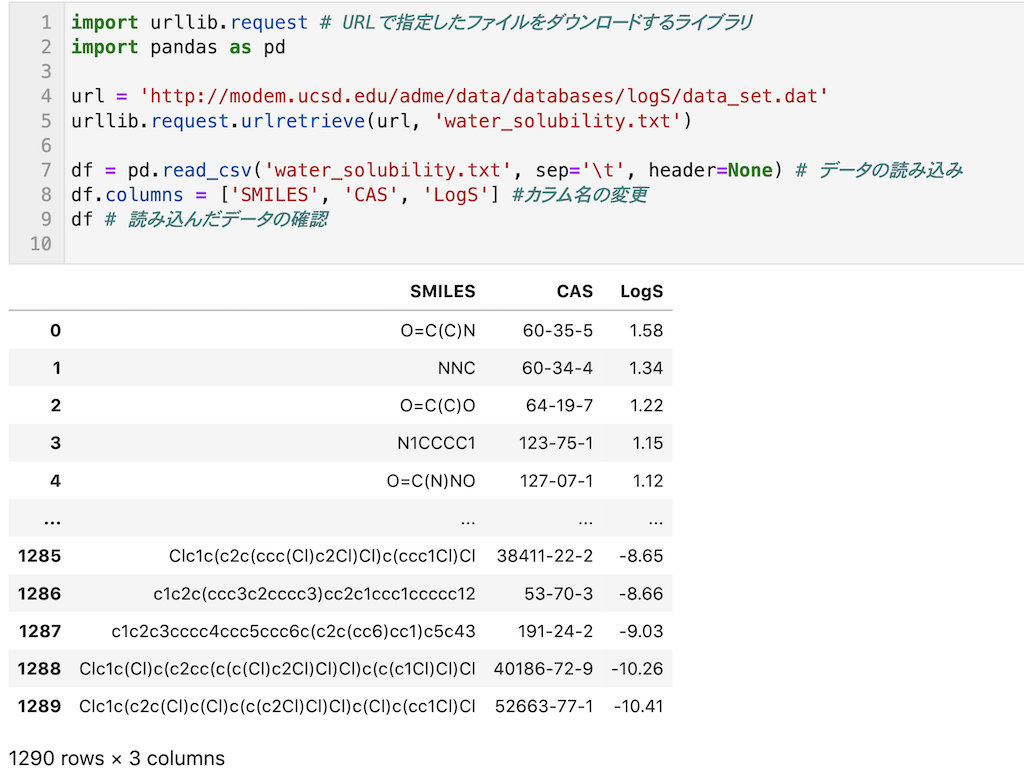
下記のコードでデータをダウンロードし、データをdfとします。
1290個の化学構造(SMILES表記)と水溶解度(Log10S)が格納されています。
(下記のコードは覚える必要は全くないです。コピペして使ってください)
import urllib.request # URLで指定したファイルをダウンロードするライブラリ
import pandas as pd
url = 'http://modem.ucsd.edu/adme/data/databases/logS/data_set.dat'
urllib.request.urlretrieve(url, 'water_solubility.txt')
df = pd.read_csv('water_solubility.txt', sep='\t', header=None) # データの読み込み
df.columns = ['SMILES', 'CAS', 'LogS'] #カラム名の変更
df # 読み込んだデータの確認
データを読み込めたので、早速「SMILES→molファイルへの変換」を実施します。
SMILES→molファイルへの変換
SMILES→molファイルに変換する前に、molファイルに変換できないデータを削除します。
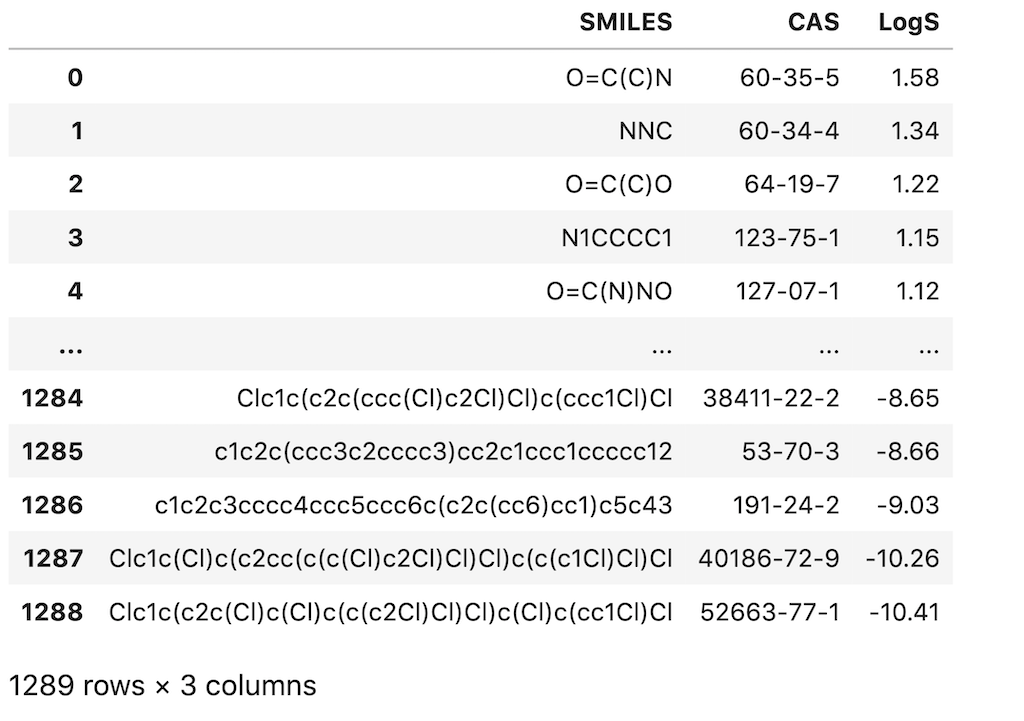
下記コードでは、for文を使ってdfの’SMILES’列から一つずつSMILESを取り出して、Chem.MolFromSmiles(smile)でmolファイルに変換し、mols_listに格納しています。
この時、molファイルに変換できないデータもある可能性があるので、変換できないものは欠損値(np.nan)としてmols_listに格納しておきます。molファイルに変換できないデータは使えないのでdfから削除しています。dfが1290個から1289個になっていることが確認できます。
#smiles→molsファイルの変換
import numpy as np
from rdkit import Chem
mols_list = []
for smile in df['SMILES']:
try:
mols_list.append(Chem.MolFromSmiles(smile))
except:
mols_list.append(np.nan)
#molファイルに変換できなかったSMILESの行番号を確認しdfから削除
drop_index = pd.Series(mols_list)[pd.Series(mols_list).isnull()].index.to_list()
df = df.drop(index = drop_index).reset_index(drop = True)
SMILES→molファイルに変換できないデータは削除したので、molファイルに変換します。
変換できないデータは全て削除しているので、全てのSMILESがmolファイルに変換されています。
mols_listの中に1289個のmolファイルが格納されています。
from rdkit import Chem
mols_list = []
for smile in df['SMILES']:
try:
mols_list.append(Chem.MolFromSmiles(smile))
except:
mols_list.append(np.nan)
「SMILES→molファイルへの変換」が完了したので、続いて「molファイル→ベクトルへの変換」を実施します。
molファイル→ベクトルへの変換
今回はrdkit記述子を使って「molファイル→ベクトルへ変換」します。
下記のコードでは、rdkit記述子で208次元に変換されたベクトルをX_trainに格納しています。
また今回は欠損値はなかったため、このまま計算に使用しています。
1289個の化合物が全て208次元のベクトルに変換されていることが確認できます。
(コードは覚える必要はなく、こんな感じで書くんだくらいの理解度でOKです)
from rdkit.Chem import Descriptors from rdkit.ML.Descriptors import MoleculeDescriptors descriptor_names = [descriptor_name[0] for descriptor_name in Descriptors._descList] descriptor_calculator = MoleculeDescriptors.MolecularDescriptorCalculator(descriptor_names) rdkit_descriptors_results = [descriptor_calculator.CalcDescriptors(mol) for mol in mols_list] X_train = pd.DataFrame(rdkit_descriptors_results, columns = descriptor_names) X_train
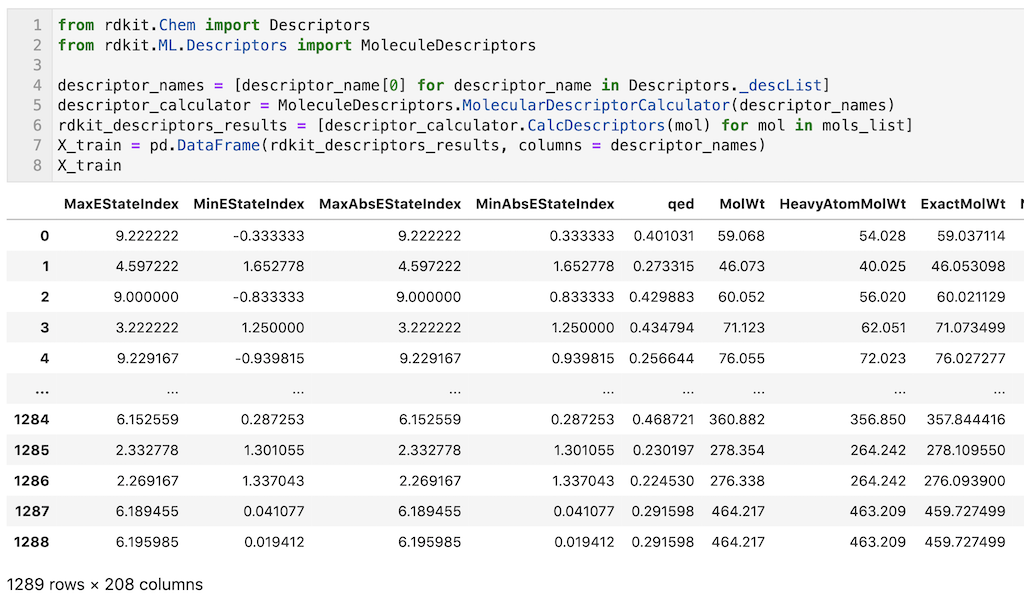
計算のため、上で算出したX_trainとy_trainを結合してdf_trainとします。
y_train = df['LogS'] df_train = pd.concat([X_train,y_train], axis = 1)
それではデータの前処理が終わったので学習を実施し、予測精度の確認をしていきます!
学習と予測精度の確認
今回はlightgbmを使ってモデル構築します。
精度検証はKFoldを使って10分割交差検証を実施しています。結果は、y_test(正解データ)とy_pred(モデルの予測結果)をverificationに格納しています。
verificationを出力すると、y_testとy_predが格納されていることがわかります。
from sklearn.model_selection import KFold
import lightgbm as lgb
verification = pd.DataFrame()
verification['y_test'] = df['LogS']
#KFoldの設定
kf = KFold(n_splits = 10, shuffle = True)
#交差検証
for train_idx, test_idx in kf.split(df):
X_train_ = df_train.iloc[train_idx, :-1]
y_train_ = df_train.iloc[train_idx, -1]
X_test = df_train.iloc[test_idx, :-1]
y_test = df_train.iloc[test_idx, -1]
model = lgb.LGBMRegressor()
model.fit(X_train_, y_train_)
verification.loc[test_idx, 'y_pred'] = model.predict(X_test)
verification
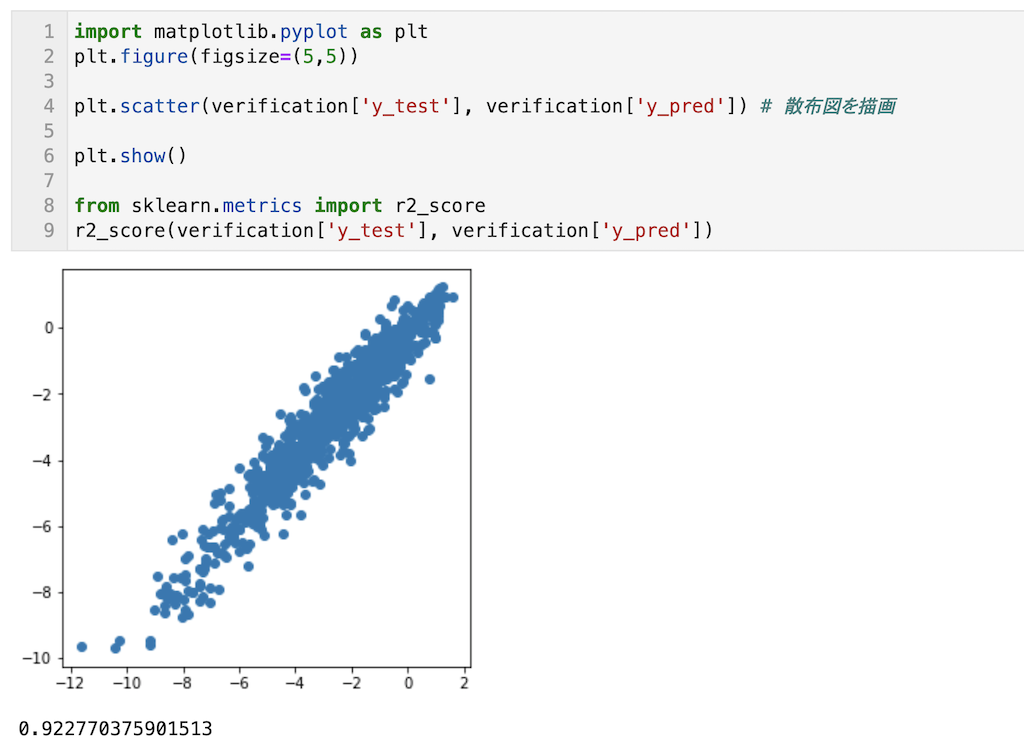
最後に予測精度結果を下記に示します。
グラフから全てのyの値で高い予測精度であることが確認できます。
またr2_scoreは0.92と非常に高い予測精度である事が確認できます。
ベクトル変換の手法を変えたり、使用する機械学習モデルを変えることで、もう少し予測精度をあげれると思います。
import matplotlib.pyplot as plt plt.figure(figsize=(5,5)) plt.scatter(verification['y_test'], verification['y_pred']) # 散布図を描画 plt.show() from sklearn.metrics import r2_score r2_score(verification['y_test'], verification['y_pred'])
今回の記事はここまでです。
pythonを活用したケモインフォマティクスではrdkitはマストなので、是非この記事で使い方をマスターしてください。
✅ 仕事(研究開発)で機械学習を活用したい・し始めた人は下記記事もお勧め
この度、『仕事で活用するための化学系データサイエンス入門』を執筆しました!
仕事で4年・1人で機械学習を活用した経験、予測を試す(実験)ところまで全て自分で実施した経験を基にリアルを書きました。

また下記記事では、化学の研究開発で機械学習を活用する意義を深掘りしてます。
「機械学習を活用する意義」を周囲に正確に説明できないと、仕事で機械学習を活用できません。
仕事で機械学習を活用する際の、最初の関門が「周囲への説明」なので、是非読んで欲しいです。

そして皆さんの機械学習の勉強がさらに進み、理解が深まることを願っています。

