

皆さん、こんにちは。ノムオです。
突然ですが、こんな悩みを抱えていないでしょうか。
・1個の初期構造だけで構造最適化していいのかわからない😭
・より安定な構造を見逃していないか不安😭
・RDKitで複数のコンフォマーを生成する方法がわからない😭
今回の記事では、「RDKitで複数のコンフォマーを生成する理由とそのやり方」について書いていきたいと思います。
構造最適化計算の流れについては下記記事を参考にしてください!

早速書いていきます。
なぜ構造最適化計算では複数のコンフォマーを生成するのか
一言で言うと、
「1つの初期構造だけで構造最適化すると、より安定な構造を見逃す可能性がある」
から。
(構造最適化計算は初期構造の近傍にある局所最適解へ収束しやすいため、1つの初期構造だけでは、より低エネルギーな構造を見逃す可能性があるという話です。)
そのため、「初期構造を複数生成(複数のコンフォマーを生成) → 構造最適化する」ことで、より安定な構造を探索しようという考え方です。
今回の記事では、イブプロフェンを例にして、「複数のコンフォマーを生成 → 構造最適化」を実施し、それぞれのエネルギーの比較を実施したいと思います。
(複数のコンフォマーを生成する意義を考えます)
RDKitで複数のコンフォマーを生成する方法
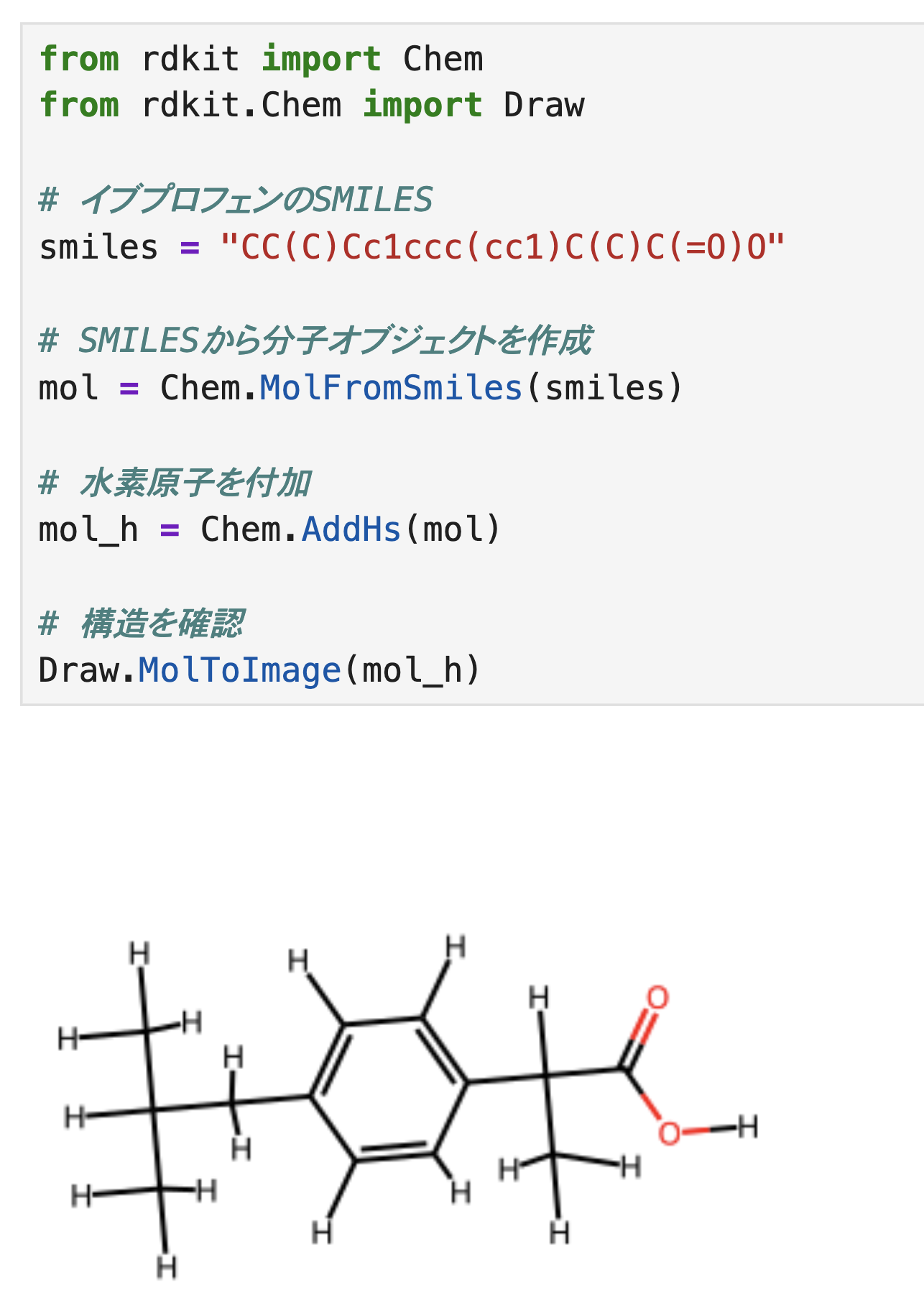
まずはイブプロフェンの分子を生成し、水素を生やします。
①SMILES → 分子作成
from rdkit import Chem from rdkit.Chem import Draw # イブプロフェンのSMILES smiles = "CC(C)Cc1ccc(cc1)C(C)C(=O)O" # SMILESから分子オブジェクトを作成 mol = Chem.MolFromSmiles(smiles) # 水素原子を付加 mol_h = Chem.AddHs(mol) # 構造を確認 Draw.MolToImage(mol_h)
準備ができたので、ここから「複数のコンフォマーを生成 → 構造最適化」し、エネルギーを比較してみます。
複数のコンフォマーを生成
複数のコンフォマーの生成には、RDKitのAllChem.EmbedMultipleConfs()関数を使います。
下記コードの「AllChem.EmbedMultipleConfs()」の ( ) の中身ですが、
・mol_h → 対象分子(今回は水素を付加したイブプロフェン)
・numConfs=20 → 生成するコンフォマー数
・params=params →どんな条件・方法で3次元構造を作るか
を指定しています。
またこの関数の出力(conf_ids)は、生成されたコンフォマーそのものではなく、生成された各コンフォマーを識別するためのID一覧が格納されます。
一方、生成された3次元構造そのものは「mol_h」の中に保存されています。
from rdkit.Chem import AllChem
# ETKDGv3の設定
params = AllChem.ETKDGv3()
params.randomSeed = 42
# 複数コンフォマーを生成
conf_ids = AllChem.EmbedMultipleConfs(
mol_h,
numConfs=20,
params=params
)
print(f"生成されたコンフォマー数: {len(conf_ids)}")
print(list(conf_ids))

出力を見てみると20個のIDができていることが確認できます。
複数のコンフォマーが生成できたので、MMFFで構造最適化します。
MMFFによる複数のコンフォマーの構造最適化
生成した20個のコンフォマーを、RDKitのAllChem.MMFFOptimizeMoleculeConfs( )関数を使って構造最適化します。
この関数を実行すると、mol_hの中に保存されている全てのコンフォマーをまとめてMMFFで最適化できます。
numThreads=0はこのPCで使える最大数のCPUスレッドを使って計算するという意味です。
# 各コンフォマーをMMFFで構造最適化
results = AllChem.MMFFOptimizeMoleculeConfs(
mol_h,
numThreads=0
)
# 各コンフォマーのエネルギーを表示
for conf_id, result in zip(conf_ids, results):
print(conf_id, result)
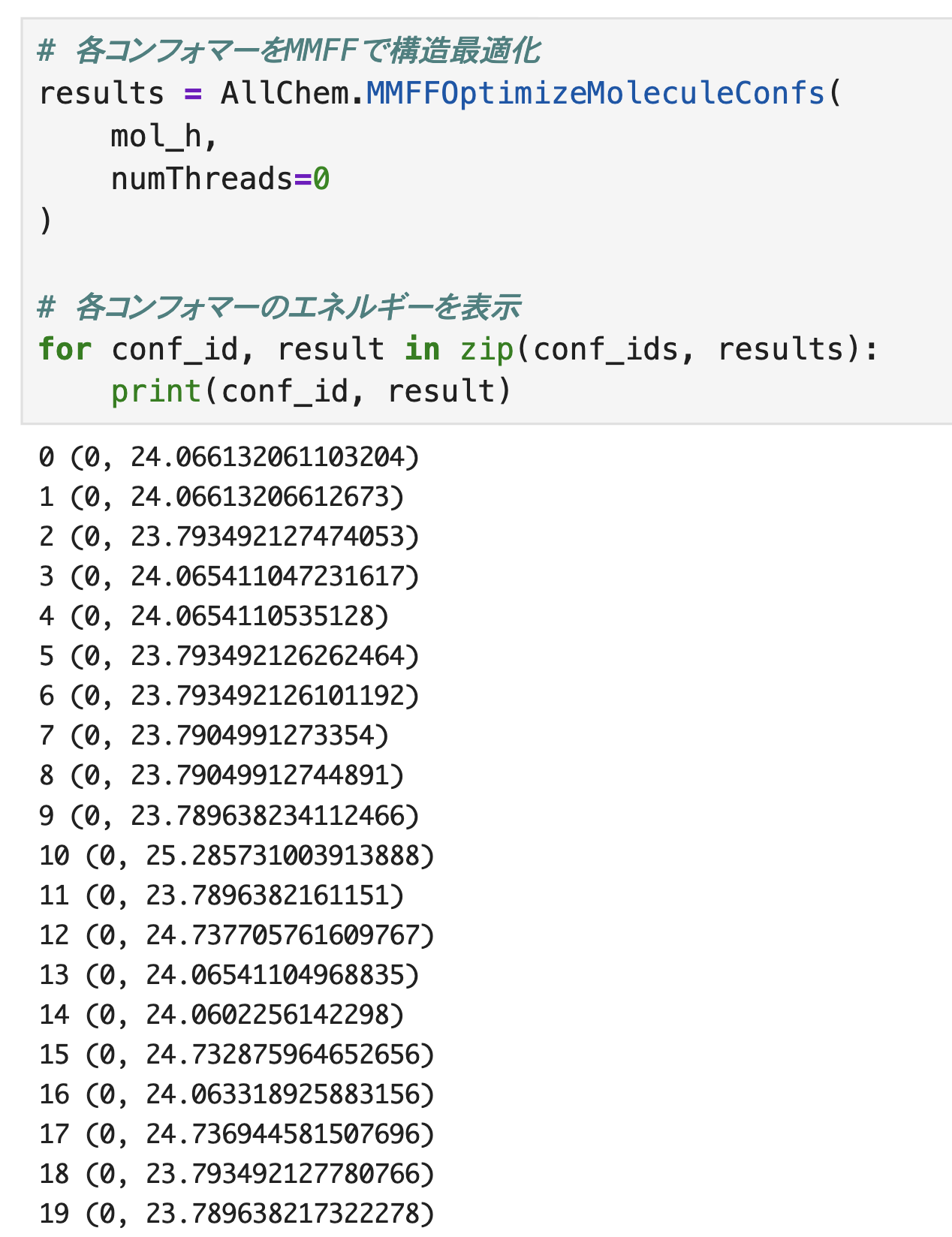
出力結果の見方ですが、
・1番左の0,1,2・・・ → コンフォマーID
・( )内の 0 → 最適化が収束したことを示す
・( )内の「23.・・・」 → 最適化後のエネルギー
MMFFによる構造最適化後のエネルギーが異なることが確認できます。
またエネルギーだけ見てもよくわからないので、最後にpy3Dmolを使って、エネルギーの最も低かったコンフォマーを可視化してみます。
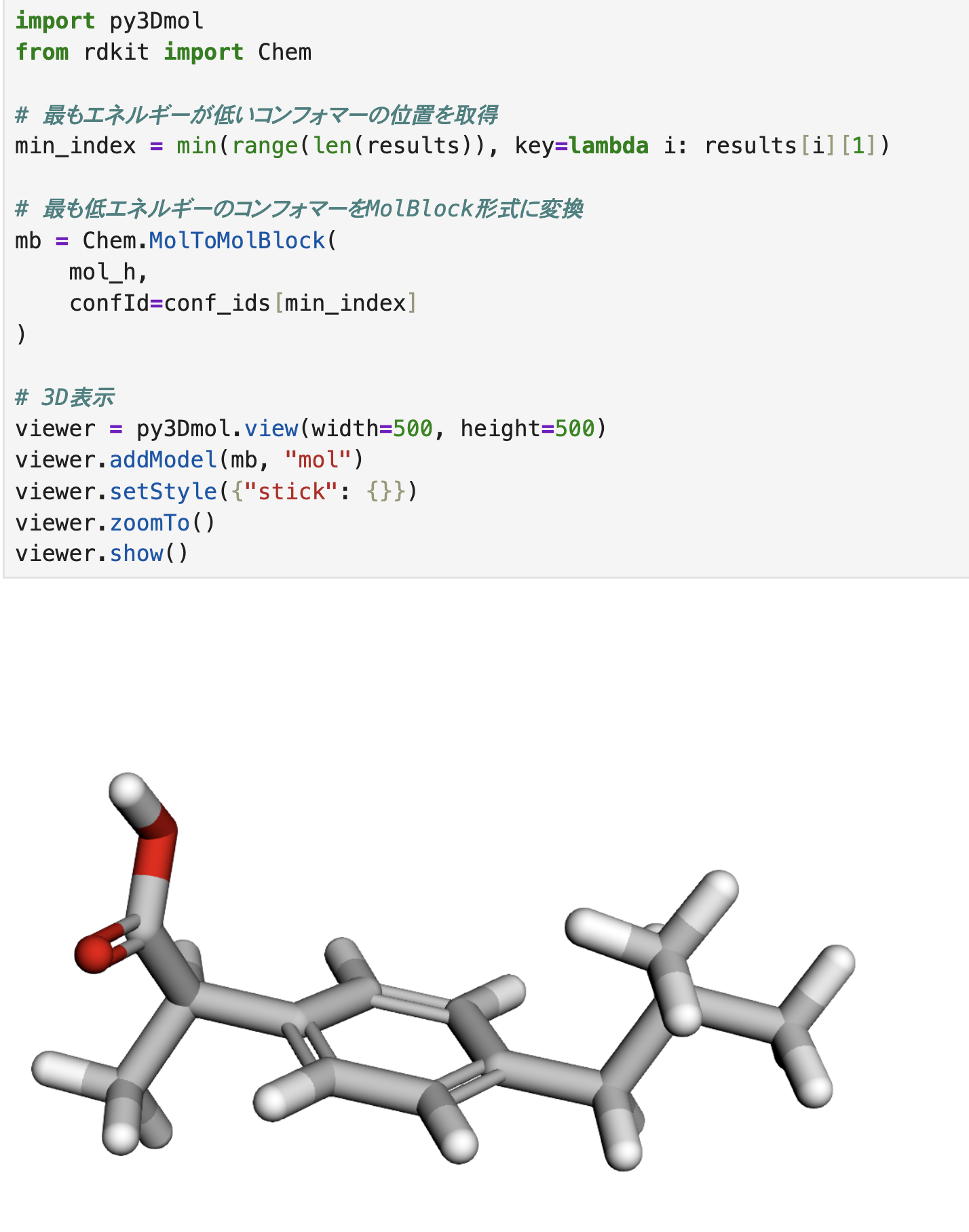
最適化された構造の可視化
RDKitのChem.MolToMolBlock( )関数を使って、molブロック形式に変換(mbという変数名で保存)し、それを使ってpy3Dmolで可視化しています。
今回は、resultsの中から最もエネルギーが低いコンフォマーを取得し、その構造を表示しています。
import py3Dmol
from rdkit import Chem
# 最もエネルギーが低いコンフォマーの位置を取得
min_index = min(range(len(results)), key=lambda i: results[i][1])
# 最も低エネルギーのコンフォマーをMolBlock形式に変換
mb = Chem.MolToMolBlock(
mol_h,
confId=conf_ids[min_index]
)
# 3D表示
viewer = py3Dmol.view(width=500, height=500)
viewer.addModel(mb, "mol")
viewer.setStyle({"stick": {}})
viewer.zoomTo()
viewer.show()
今回の記事では、RDKitを使って複数のコンフォマーを生成し、MMFFによる構造最適化とエネルギー比較を実施しました。
構造最適化では、1つの初期構造だけを使うと、より安定な構造候補を見逃す可能性があります。
そのため、「複数のコンフォマーを生成 → それぞれ構造最適化 → エネルギーを比較」という流れで計算することで、より低エネルギーな構造候補を探索しやすくなります。
特に、分子の3D構造を使った記述子計算、立体配座の比較、分子間相互作用の検討などを行う場合には、コンフォマーを意識することが重要です。
今回の記事が、RDKitで複数のコンフォマーを扱う際の参考になれば幸いです。
ノムオ
