

皆さんこんにちは。ノムオです。
突然ですが、こんな悩みを抱えていないでしょうか。
・化学構造の構造最適化計算のやり方がわからない😭
・構造最適化計算の手法はいっぱいあるけど、結局どれがいいのかわからない😭
今回の記事では、「rdkitを活用した化学構造の構造最適化計算」について書いていきたいと思います。
カフェインを例にしてrdkitを活用して構造最適化計算をやっていきたいと思います。
早速書いていきます。
構造最適化計算とは
構造最適化計算とは、計算により、分子のエネルギーが低くなるように原子の位置を調整し、安定な構造を求めることです。
化合物はとりうる配座によってエネルギーが異なります。
現実世界ではエネルギーが低い構造ほど安定に存在しやすいため、構造最適化によって安定構造を求めることで、実際の分子の状態に近い構造で計算できます。
rdkitでは一般的に以下の流れで構造最適化を行います。
①SMILES → 分子作成
②水素付加
③ETKDGで初期構造生成
④UFFやMMFFで構造最適化
分子から初期構造を生成し、その後、構造最適化するという流れです。
基本的には構造最適化はこの流れで実施しますが、④の構造最適化は目的(どの程度、構造最適化したいか)に応じて、手法選択が必要になります。
化学記述子の算出であればUFFやMMFFで最適化するくらいでいいですし、化学反応を取り扱うような系ではxTBやDFTによる構造最適化が必要になります。
計算コストと目的次第という話です。
それではカフェインを例に構造最適化計算(①〜④)を実践してみます。
構造最適化計算の実践
まずはカフェインの分子生成からです。
①SMILES → 分子作成
# カフェインのSMILES smiles = "Cn1cnc2c1c(=O)n(C)c(=O)n2C" # SMILESから分子オブジェクトを作成 mol = Chem.MolFromSmiles(smiles) # 構造を確認 Draw.MolToImage(mol)
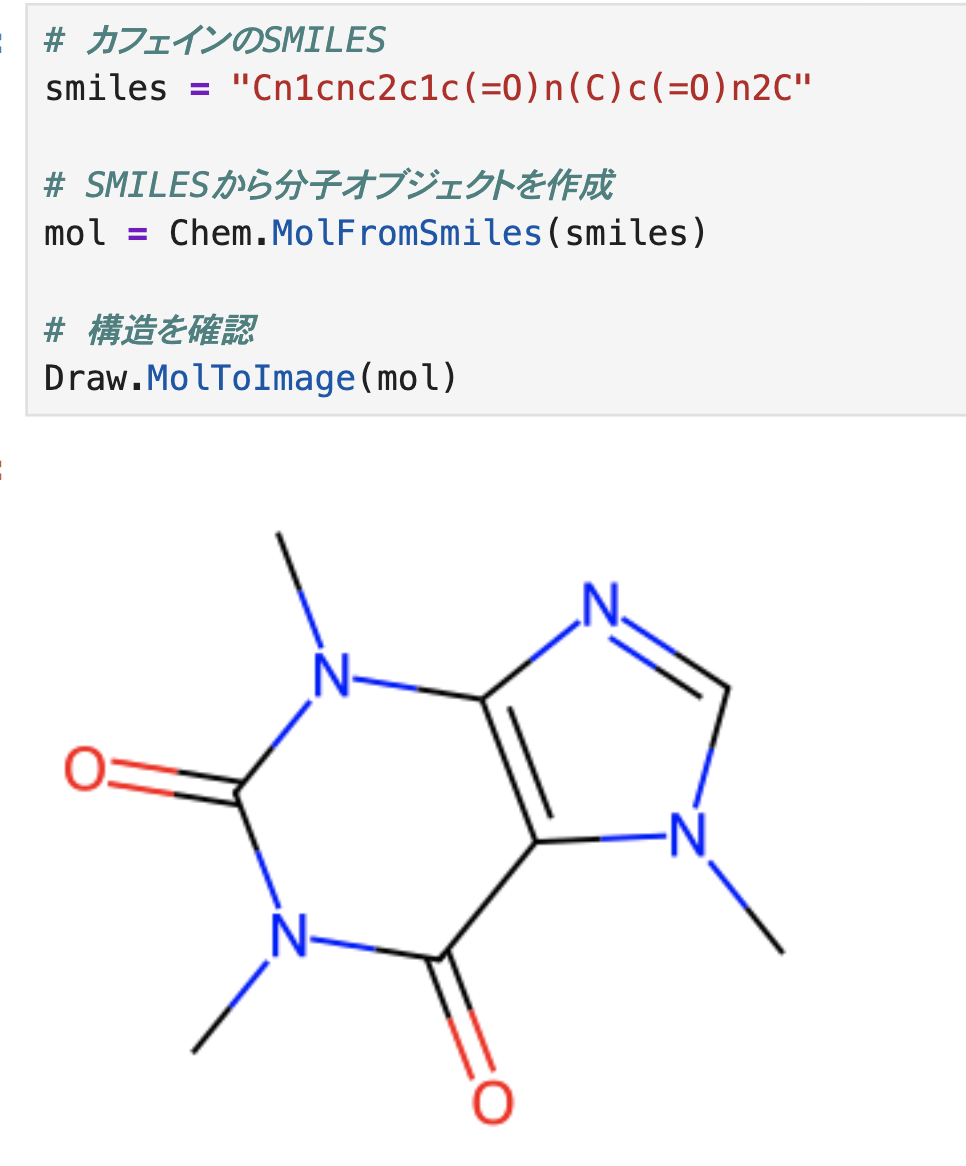
続いて生成した構造に水素を生やします。
②水素を生やす
下記コードで水素を生やすことができます。
出力結果を見ると、Hが表示されていることが確認できます。
# 水素原子を付加 mol_h = Chem.AddHs(mol) # 構造を確認 Draw.MolToImage(mol_h)
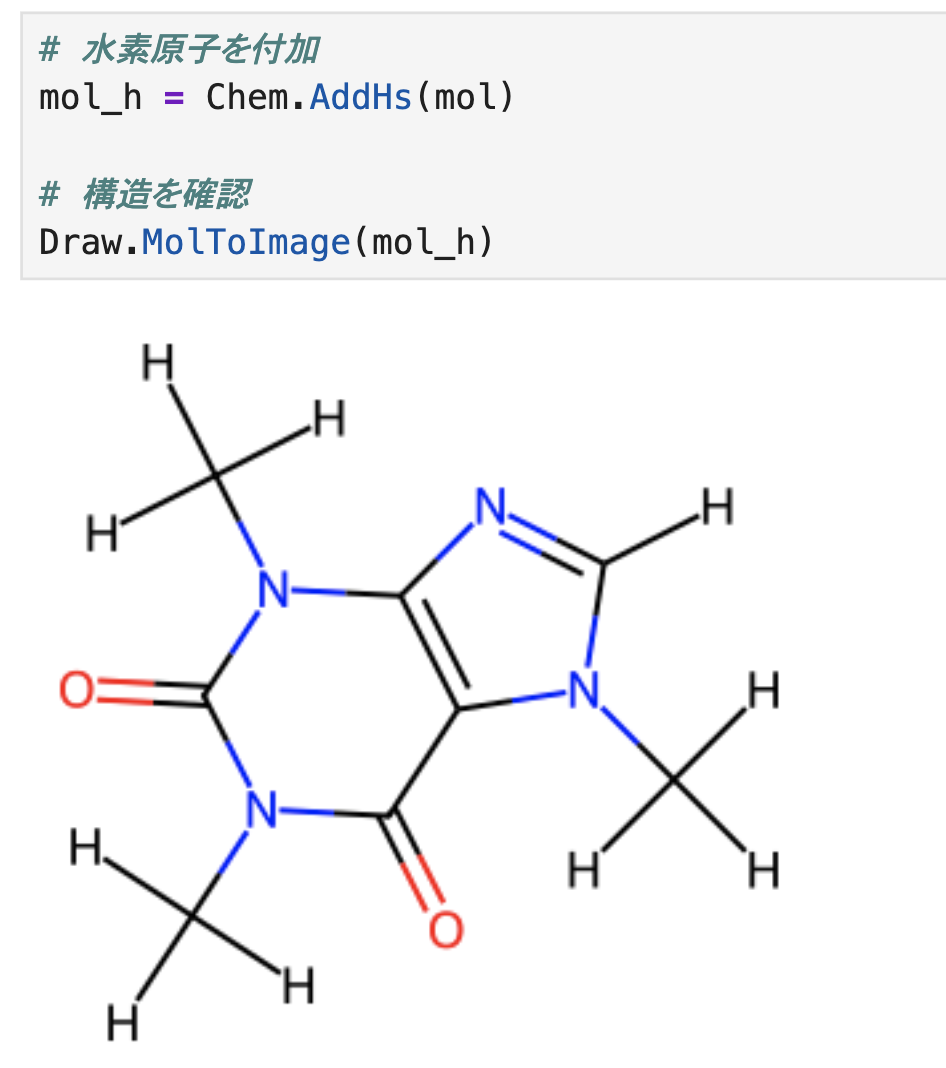
一方でHを生やしただけだと、3D座標を持っていません。
そこで③ETKDG(EmbedMolecule)を使って、構造最適化前の初期構造(各原子に3D座標を割り当てる)を行います。
③ETKDGによる初期構造生成
下記コードで初期構造を生成できます。
今回は解説のためにrandomSeedを固定してます。randomSeedを固定することで同じ初期構造を作ることができます。
(なおrandomSeedを選択しなければ違う初期構造を生成できます。)
また出力結果が0というのは3D構造(初期構造)の生成が正常に終了したという意味です。
# ETKDG法の設定 params = AllChem.ETKDGv3() params.randomSeed = 42 # 3D構造を生成 AllChem.EmbedMolecule(mol_h, params)
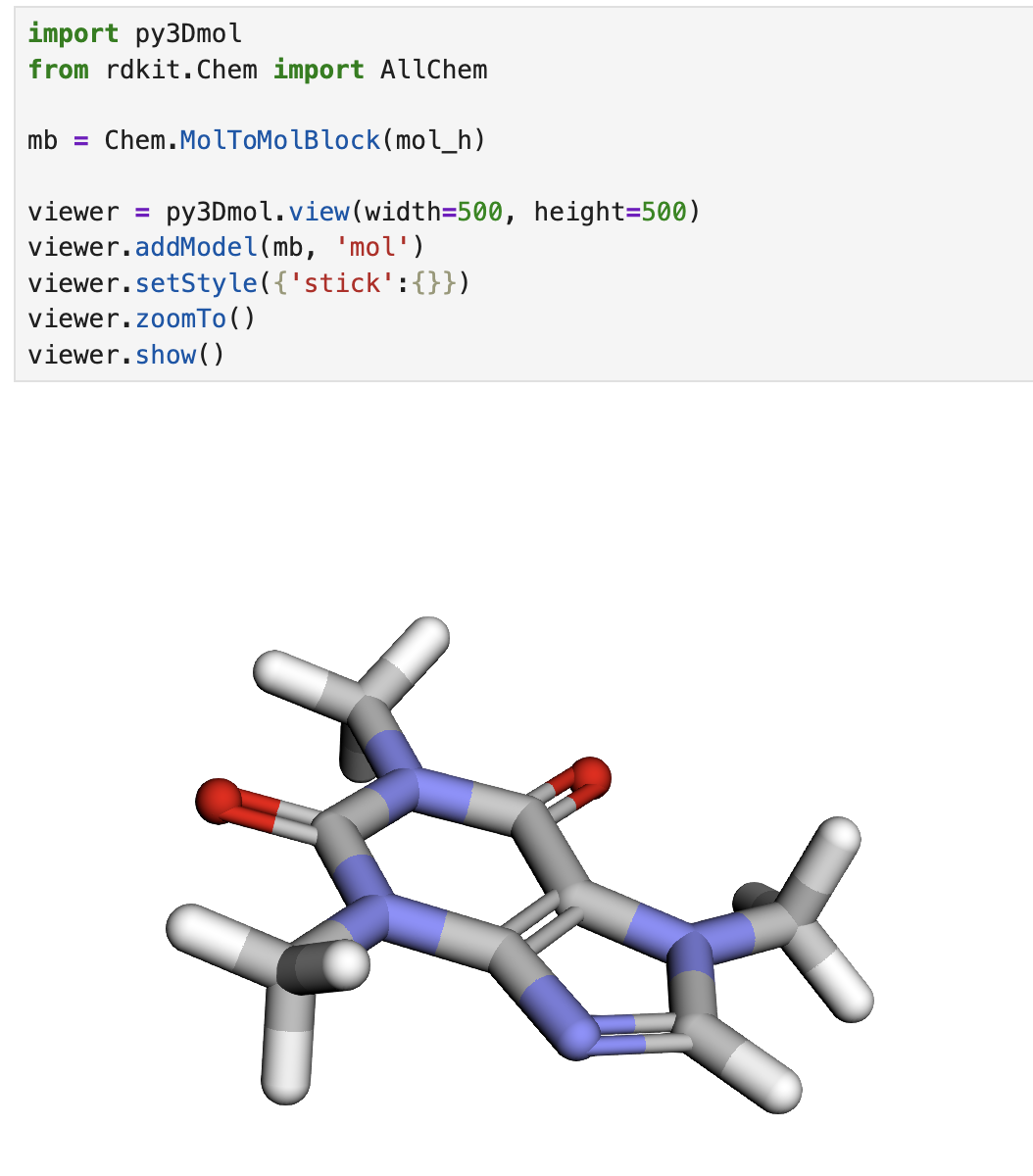
出力結果が0だけだと、どんな初期構造ができたのかイメージできないので、py3Dmolを使って、3D構造が生成できていることを確認してみます。
しっかり3D構造が生成できていることが確認できました。
import py3Dmol
from rdkit.Chem import AllChem
mb = Chem.MolToMolBlock(mol_h)
viewer = py3Dmol.view(width=500, height=500)
viewer.addModel(mb, 'mol')
viewer.setStyle({'stick':{}})
viewer.zoomTo()
viewer.show()
初期構造ができたので、構造最適化計算を実施します。
④MMFFで構造最適化
③で生成した初期構造からMMFFで構造最適化を実施します。
出力結果が0というのは構造最適化が正常に終了したという意味です。
from rdkit.Chem import AllChem # MMFFで構造最適化 result = AllChem.MMFFOptimizeMolecule(mol_h) print(result)

出力結果だけみてもイメージが湧かないので、構造最適化後の3D構造をpy3Dmolを使って可視化してみます。
import py3Dmol
from rdkit import Chem
# MMFF最適化後の mol_h をmolブロック形式に変換
mb = Chem.MolToMolBlock(mol_h)
# 3D表示
viewer = py3Dmol.view(width=500, height=500)
viewer.addModel(mb, "mol")
viewer.setStyle({"stick": {}})
viewer.zoomTo()
viewer.show()
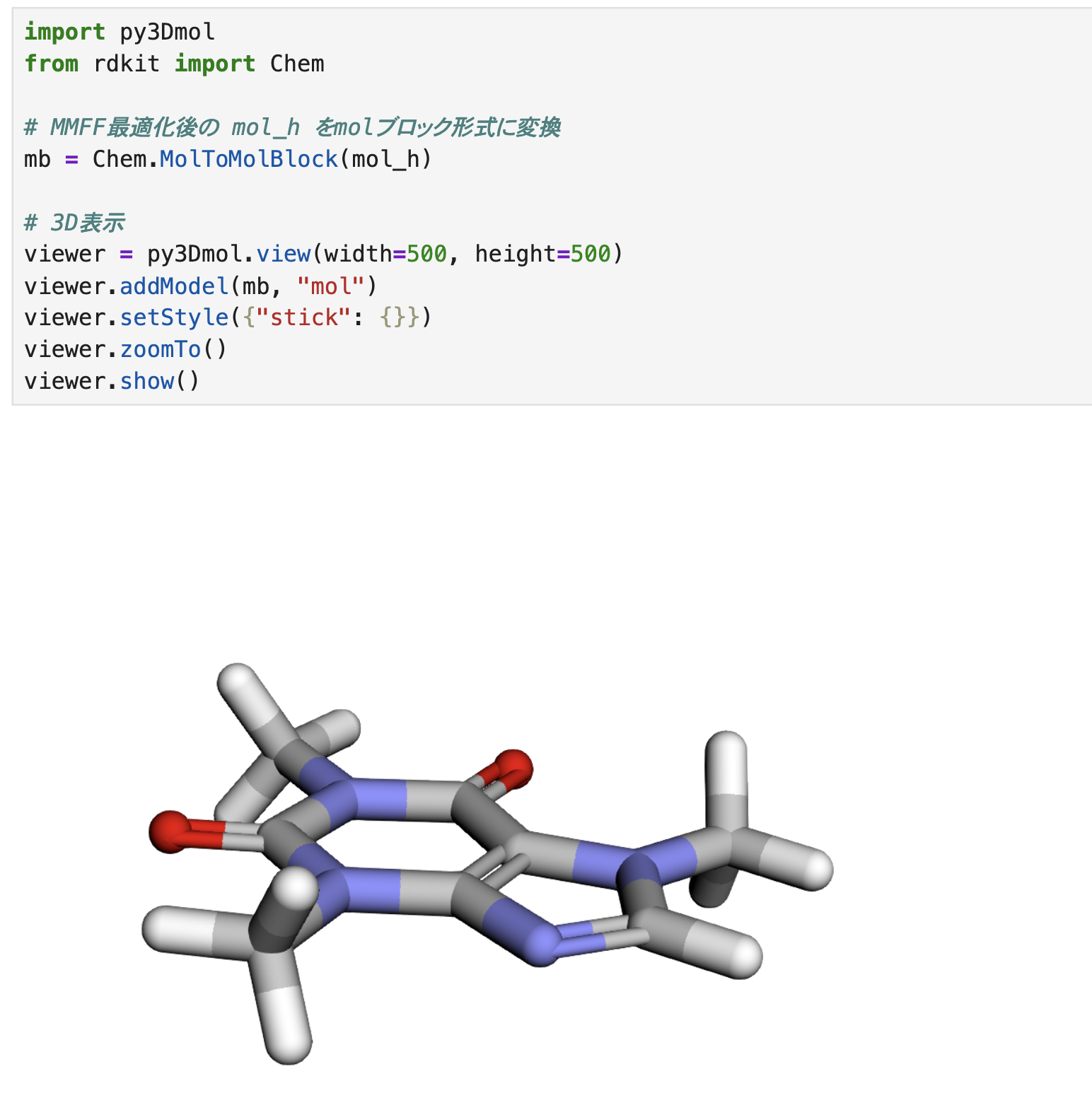
カフェインは環構造を有しており、平面性が高く、「構造最適化前後で構造がどう変わったのか?」イメージするのが難しいので、最後に、MMFFによる構造最適化でエネルギーがどの程度低下したか確認してみます。
構造最適化計算により約14kcal/mol安定な構造を出力できたことが確認できました。
(なお、このエネルギーの絶対値自体には大きな意味はなく、同じ分子内で構造最適化前後の差を比較することが重要です。)
from rdkit import Chem
from rdkit.Chem import AllChem
# カフェイン
smiles = "Cn1cnc2c1c(=O)n(C)c(=O)n2C"
# 分子作成
mol = Chem.MolFromSmiles(smiles)
mol_h = Chem.AddHs(mol)
# ETKDGで3D構造生成
params = AllChem.ETKDGv3()
params.randomSeed = 42
AllChem.EmbedMolecule(mol_h, params)
# 最適化前エネルギー
props = AllChem.MMFFGetMoleculeProperties(mol_h)
ff = AllChem.MMFFGetMoleculeForceField(mol_h, props)
energy_before = ff.CalcEnergy()
# MMFF最適化
AllChem.MMFFOptimizeMolecule(mol_h)
# 最適化後エネルギー
ff = AllChem.MMFFGetMoleculeForceField(mol_h, props)
energy_after = ff.CalcEnergy()
print(f"最適化前: {energy_before:.2f} kcal/mol")
print(f"最適化後: {energy_after:.2f} kcal/mol")
print(f"変化量 : {energy_after-energy_before:.2f} kcal/mol")

今回の記事は以上です。
ケモインフォマティクスでは化学構造の取り扱いはほぼ必須です。
その中で構造最適化計算は確実に実施することになりますので、今回の記事が参考になれば幸いです。
ノムオ

